
Mitochondrial Dysfunction and Chronic Diseases: A Mechanistic Review with Lifestyle Recommendations
-
摘要: 探讨了线粒体功能障碍在慢性疾病发病机制中的关键作用,包括代谢性疾病、神经退行性疾病、心血管疾病和癌症,重点关注其对ATP生成、活性氧(ROS)产生和细胞稳态的影响。同时也阐述了驱动疾病进展的机制性关联,如受损的线粒体生物合成和过度的氧化应激。在应用层面,本文提供了基于实证的生活方式干预措施,特别是饮食策略(如生酮饮食、植物源营养、间歇性禁食和热量限制),这些干预通过促进线粒体生物合成与减少氧化损伤来增强线粒体功能。此外,评估了耐力训练、高强度间歇训练和抗阻训练等运动方式通过多种信号通路刺激线粒体适应能力。本文强调以能量为核心的健康理念,即优先考虑线粒体能量生产以促进整体健康。通过整合细胞、动物和人体层面研究,突出饮食与运动策略的协同效益,并为预防和管理慢性疾病提供切实可行的建议。Abstract: This review explores the critical role of mitochondrial dysfunction in the pathogenesis of chronic diseases, including metabolic disorders, neurodegenerative diseases, cardiovascular conditions, and cancer, emphasizing its impact on ATP production, reactive oxygen species generation, and cellular homeostasis. It elucidates mechanistic links, such as impaired mitochondrial biogenesis and excessive oxidative stress, that drive disease progression. The manuscript evaluates evidence-based lifestyle interventions, specifically dietary strategies like ketogenic diets, plant-based nutrition, intermittent fasting, and caloric restriction, which enhance mitochondrial function by promoting biogenesis and reducing oxidative damage. Additionally, exercise modalities, including endurance training, high-intensity interval training, and resistance training, are assessed for their ability to stimulate mitochondrial adaptations via various signaling pathways. By emphasizing energy-centric health, which prioritizes mitochondrial energy production for overall well-being, this review integrates cellular, animal, and human studies to underscore the synergistic benefits of combined dietary and exercise strategies, providing actionable insights for preventing and managing chronic diseases.
-
Keywords:
- mitochondrial /
- chronic diseases /
- dysfunction /
- metabolic disorders
-
线粒体是双层膜结构的细胞器,作为细胞能量生产的主要场所,通过氧化磷酸化发挥作用。这些细胞器在生成三磷酸腺苷(Adenosine Triphosphate, ATP)中发挥基础作用,ATP是维持细胞功能和稳态所必需的能量“货币”[1]。线粒体通过三羧酸循环(Tricarboxylic Acid, TCA)代谢碳水化合物、脂肪和蛋白质的底物,为电子传递链提供电子以合成ATP[2]。由于其高能量需求,骨骼肌、心脏和大脑等组织的线粒体密度较高,凸显了其在维持生理过程中的重要性[3]。除了能量代谢外,线粒体还调节关键的细胞功能,包括活性氧(Reactive Oxygen Species, ROS)的产生、钙稳态和细胞凋亡,因此其完整性对整体健康至关重要[4]。
已有研究证实,线粒体功能障碍与多种慢性疾病的发病机制相关,包括代谢性疾病、心血管疾病和神经退行性疾病[5]。线粒体效率受损、ROS过度产生以及线粒体生物合成失调导致肥胖、胰岛素抵抗、2型糖尿病(Type 2 Diabetes, T2D)和心血管疾病(图1)[6]。多种细胞紊乱导致线粒体功能障碍,在代谢性疾病的发展中起核心作用。过量的ROS、ATP合成减少、线粒体动力学失调、线粒体生物合成不良和线粒体基因表达低下等因素会损害线粒体功能。这种功能障碍破坏了细胞的能量平衡,导致肥胖、T2D、血脂异常和心血管疾病等代谢性疾病的发生。越来越多的证据表明线粒体功能障碍是慢性疾病病理的关键中介因素,而最新研究则显示,靶向干预措施,特别是饮食调整和体育活动,对恢复线粒体健康至关重要。热量限制(Caloric Restriction, CR)、生酮饮食(Ketogenic Diet, KD)和靶向微量营养素补充等营养策略已被证实具有增强线粒体生物合成和效率的潜力[7]。同样,运动尤其是耐力训练和高强度间歇训练(High-Intensity Interval Training, HIIT),被认为是线粒体适应的强效刺激因素,其作用机制主要通过上调线粒体生物合成实现[8]。
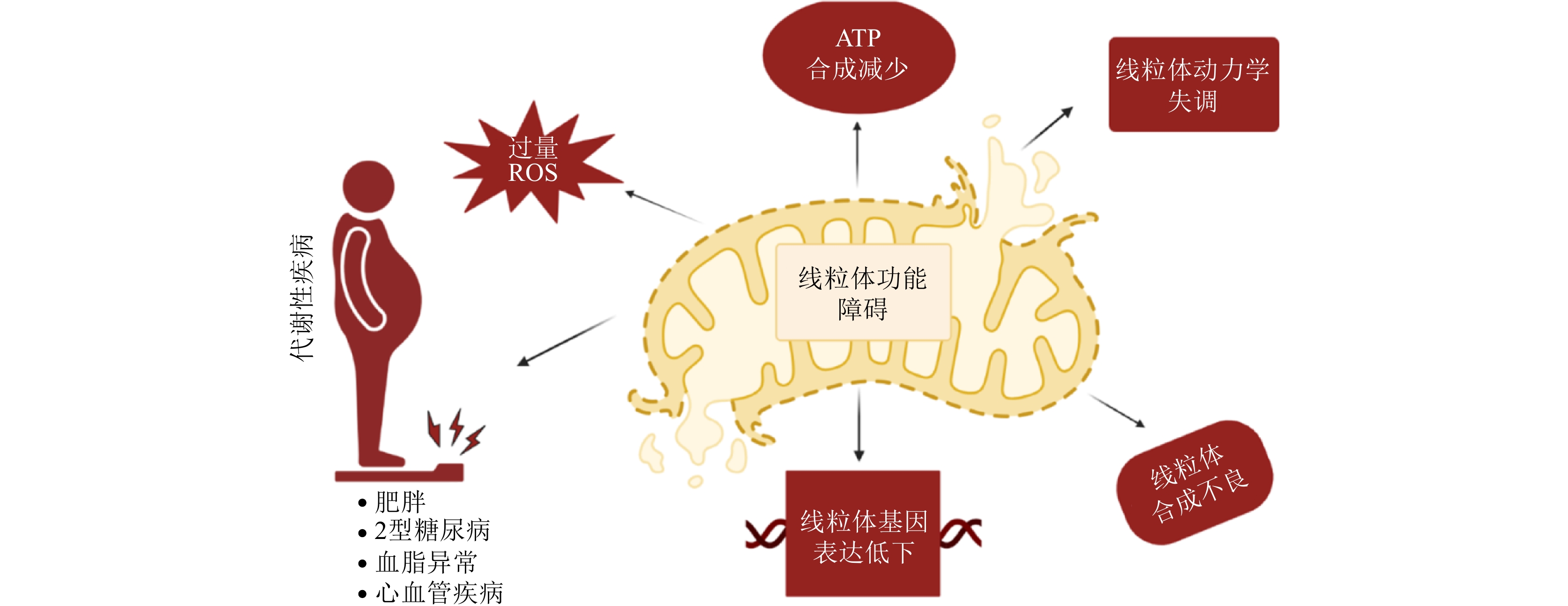 图 1 多种细胞紊乱导致线粒体功能障碍在代谢性疾病的发展中起核心作用Fig. 1 Various cellular disturbances contribute to mitochondrial dysfunction which plays a central role in the development of metabolic diseases
图 1 多种细胞紊乱导致线粒体功能障碍在代谢性疾病的发展中起核心作用Fig. 1 Various cellular disturbances contribute to mitochondrial dysfunction which plays a central role in the development of metabolic diseases 下载:
全尺寸图片
下载:
全尺寸图片
本文将讨论线粒体功能障碍与慢性疾病之间的机制关系,提供基于证据的营养及运动策略,旨在帮助线粒体功能恢复,并进一步改善健康状态。具体而言,将探讨饮食策略如何增强线粒体生物合成和功能,如CR、KD和特定微量营养素补充,并将探讨体育活动(包括耐力训练和抗阻训练)对线粒体适应和代谢健康的影响。通过整合细胞、动物和人类研究证据,强调生活方式干预在缓解线粒体功能障碍中的重要性,为开发针对慢性疾病的预防和治疗策略提供实用见解。
1. 线粒体功能障碍与慢性疾病的机制联系
线粒体生物合成受损是多种慢性疾病线粒体功能障碍的关键特征,包括代谢性疾病、神经退行性疾病、心血管疾病和癌症。线粒体生物合成过程受到转录共激活因子网络的严格调控,特别是过氧化物酶体增殖物激活受体γ共激活因子1-α(Proliferator-Activated Receptor Gamma Coactivator 1-Alpha, PGC-1α),它协调线粒体结构和功能编码所必需的蛋白质核基因和线粒体基因的表达[9]。在肥胖状态下,PGC-1α活性降低(通常由炎症、氧化应激或营养过剩引发)会导致线粒体含量减少和能量生产能力下降[10]。这种下降限制了ATP的合成并加剧了细胞应激,因为较少的线粒体难以满足代谢需求,从而形成功能障碍的恶性循环,导致疾病的发展。
在T2D等代谢性疾病中,骨骼肌和胰腺β细胞的线粒体功能障碍会损害氧化磷酸化,导致ATP生成减少[11-12]。例如,在骨骼肌中ATP不足会损害胰岛素介导的葡萄糖摄取,扰乱脂质代谢,从而导致胰岛素抵抗。在胰腺β细胞中,ATP生成减少会破坏启动胰岛素分泌所需的能量平衡,使细胞更难有效应对血糖水平的上升。功能失调的线粒体产生的ROS增加会通过抑制葡萄糖转运蛋白4(Inhibiting Glucose Transporter 4, GLUT4)的转位,进一步破坏胰岛素信号传导,从而加剧高血糖[13]。同样,在肥胖和代谢综合征中,脂肪酸氧化减少和线粒体效率低下会促进异位脂质积累和脂肪组织炎症,从而导致全身代谢失调[14-15]。在非酒精性脂肪肝中,线粒体β氧化受损会导致肝细胞内脂质积累,而过度的ROS生成会驱动脂毒性和肝纤维化[16-17]。
线粒体功能障碍在神经退行性疾病中也扮演着重要角色。线粒体在神经元中尤为重要,其为神经细胞之间的通讯提供能量并帮助调节钙水平。阿尔茨海默病患者的线粒体损伤(如电子传递链酶活性降低和氧化应激增加)与β-淀粉样蛋白(Amyloid-β, Aβ)斑块和异常tau蛋白的积累有关,从而导致记忆丧失和认知能力下降[18-19]。帕金森病患者的多巴胺能神经元中的线粒体功能受损会导致能量短缺和持续的氧化应激,这使得细胞更难清除α-突触核蛋白等有毒蛋白质,并导致这些重要脑细胞的死亡[20-21]。PTEN诱导假定激酶1(PTEN-Induced Putative Kinase 1, PINK1)和E3泛素-蛋白连接酶(parkin)等蛋白质(有助于维持线粒体健康)的基因突变会干扰受损线粒体的清除,从而推动帕金森病的发展[22]。肌萎缩侧索硬化症患者线粒体氧化损伤和超氧化物歧化酶1(Superoxide Dismutase 1, SOD1)、TARDNA结合蛋白-43(TAR DNA-Binding Protein 43,TDP-43)等基因突变与神经细胞死亡和炎症有关,最终导致运动神经元进行性丧失[23-24]。
线粒体功能障碍是心血管疾病的关键驱动因素,因为心脏是依靠线粒体ATP的持续生成来实现收缩功能的。在心力衰竭(Heart Failure, HF)中,功能失调的心肌细胞表现出氧化磷酸化受损、线粒体生物合成减少和ROS生成过多,这些都会导致心脏能量耗竭和细胞凋亡[25-26]。同样,高血压患者的血管平滑肌细胞的线粒体功能障碍会导致ROS过度生成,从而破坏内皮功能并促进血管收缩[27]。此外,在动脉粥样硬化中,线粒体氧化应激会加速脂质过氧化、内皮功能障碍和巨噬细胞泡沫细胞的形成,从而推动斑块形成和血管炎症[28]。
除了代谢性疾病、神经退行性疾病和心血管疾病外,线粒体功能障碍还是癌症病理生理学的关键驱动因素,能够加快肿瘤生长、存活和治疗抵抗。癌细胞经过线粒体代谢重编程,倾向于有氧糖酵解而非氧化磷酸化,这种现象被称为“瓦氏效应”[29]。这种代谢转变是由线粒体功能障碍和线粒体DNA编码的电子传递链蛋白突变驱动的,这些突变改变了ATP的产生并增强了肿瘤的存活能力[30]。此外,线粒体动力学失调(如线粒体裂变增加和融合受损)支持肿瘤转移和凋亡抵抗[31]。线粒体ROS会导致氧化应激,并可激活核因子κB(Nuclear Factor Kappa-B, NF-κB)和低氧诱导因子-1α(Hypoxia-Inducible Factor-1α HIF-1α)等信号分子,进而调节炎症、血管生成和肿瘤进展的相关通路。此外,线粒体凋亡机制的缺陷使癌细胞能够逃避细胞死亡,从而导致多种癌症的化疗抵抗[32-33]。
简言之,线粒体功能障碍已被越来越多地认为是多种慢性疾病发生和发展的关键因素。线粒体是细胞的能量工厂,负责通过氧化磷酸化产生ATP,其功能受损会导致广泛的细胞和系统性问题。表1列出了与线粒体功能障碍相关的慢性疾病。如表1所示,线粒体功能障碍与这些疾病的关联机制通常涉及ATP生成减少、ROS过度产生、钙稳态紊乱和线粒体DNA突变的积累。虽然线粒体功能障碍并不是唯一的原因,但其往往是疾病病理的关键推进者或放大器。
表 1 与线粒体功能障碍相关的慢性疾病Table 1 Chronic diseases associated with mitochondrial dysfunction疾病类别 疾病名称 主要机制 代谢疾病 T2D 胰岛素敏感组织(如肌肉、肝脏)中的线粒体功能障碍会导致胰岛素抵抗和葡萄糖代谢紊乱 肥胖 脂肪组织和骨骼肌中的线粒体氧化能力下降,会导致脂肪堆积和代谢失衡 非酒精性脂肪肝 线粒体功能受损会破坏脂肪酸氧化过程,导致脂质在肝脏中积聚 神经退行性疾病 阿尔茨海默病 线粒体功能障碍会加剧氧化应激并损害能量代谢,进而导致神经元损伤和认知功能下降 帕金森病 线粒体复合体I的缺陷和ROS生成的增加与多巴胺能神经元的丢失有关 心血管
疾病心力衰竭 线粒体ATP生成的减少和氧化应激的加剧会损害心肌功能 动脉粥样硬化 血管细胞中的线粒体功能障碍会促进炎症和斑块形成 高血压 线粒体动力学受损和ROS的过量产生会导致血管功能障碍 其他 癌症 线粒体功能障碍可通过改变细胞代谢(如“瓦氏效应”)、增加ROS生成以及削弱细胞凋亡来促进肿瘤发生。与之相关的特定癌症包括乳腺癌、前列腺癌和结直肠癌,但线粒体的作用因癌症类型而异 慢性疲劳综合征(chronic fatigue syndrome) 有证据表明,有缺陷的线粒体能量生产可能是慢性疲劳综合征患者持续疲劳和身体机能下降的根本原因 衰老与年龄相关疾病 线粒体DNA突变和线粒体功能衰退是衰老过程的核心,会导致肌少症、骨质疏松和衰弱等年龄相关疾病 2. 以能量代谢为核心的健康理念
通过关注线粒体健康,可以针对许多慢性疾病的根本原因优化能量代谢并促进整体健康。理解线粒体功能障碍是以能量为核心的健康理念的基石,这一理念因其强调线粒体在能量生产、代谢和细胞健康中的关键作用而受到越来越多的关注[34]。以能量为核心的健康是一种整体理念,强调能量生产在维持最佳细胞功能、修复和生长中的重要性。相比之下,孤立式健康是指孤立地解决健康问题或症状的做法,通常局限于特定的医学或医疗专业领域。孤立式健康倾向于单独治疗身体的某些部分、疾病或症状,而不考虑它们之间的相互联系。这种方法通常只关注症状管理,而非解决健康问题的根本原因或影响因素。
以能量为核心的健康理念强调能量生产和调节的核心作用,特别是通过线粒体功能和细胞代谢来维持整体健康。通过理解身体如何在细胞水平生成和利用能量,个人和医疗保健提供者可以采取积极的策略来预防和管理多种疾病。均衡营养、规律体育活动和有效的压力管理等生活方式干预措施不仅能提高能量生产,还能增强线粒体效率、减少氧化应激,并进一步有助于免疫健康。这些实践使个人能够通过可持续性的改变,掌控自己的健康状态,减少对药物和医疗干预的依赖。这些益处尤其适用于T2D、肥胖、心血管疾病和代谢综合征等慢性或非传染性疾病的防治。
除了疾病预防外,以能量为核心的健康理念对大脑健康、运动表现和健康老龄化也具有重要意义。对于大脑,改善能量代谢可有助于延缓阿尔茨海默病和帕金森病等神经退行性疾病的进展;对于运动和健身领域,提高线粒体效率可以增强耐力、力量、疲劳恢复和整体表现,同时指导个性化的营养策略以支持代谢灵活性。此外,这种方法为开发靶向疗法提供了一些信息,从基于生活方式的干预到药物治疗,旨在增强线粒体生物合成并纠正能量代谢功能障碍。最终,以能量为核心的健康观为推进医疗保健和提高生活质量提供了一个全面、前瞻性的框架。
尽管越来越多的证据表明线粒体功能障碍与慢性疾病相关,但这一认识尚未渗透到主流医学实践中。例如,大多数医生尚未将线粒体视为直接的治疗靶点,也没有制定解决线粒体功能障碍的官方指南。虽然线粒体功能障碍作为许多慢性疾病发生发展的关键机制的认识已开始进入医学教育领域,但医疗保健提供者的广泛认知仍有限。尽管如此,越来越多的研究人员正积极推动这一科学领域的发展,为以能量为核心的健康理念奠定基础。
3. 增强线粒体功能的饮食方法
鉴于线粒体功能障碍在慢性疾病中的核心作用,通过营养和体育活动进行靶向干预已成为恢复线粒体功能和促进以能量为核心的健康观的有效策略。其中,多种饮食方法因其具有支持线粒体完整性和能量生产的功能而得到进一步研究。
KD以高脂肪和低碳水化合物摄入为特点,通过将细胞能量依赖从葡萄糖转变为脂肪酸氧化和酮体利用来增强线粒体氧化代谢。这种代谢适应减少了对糖酵解的依赖,从而降低了乳酸积累,并为大脑、心脏和骨骼肌等高线粒体需求组织器官提供了持续的能量供应。此外,通过减少对葡萄糖代谢的依赖,KD降低了胰岛素循环水平,提高了胰岛素敏感性,有助于缓解与葡萄糖过量相关的慢性炎症和氧化应激。KD中ROS生成减少,源于酮体的优先氧化,与葡萄糖氧化相比,酮体产生的自由基更少。此外,β-羟基丁酸等酮体作为信号分子,可上调参与线粒体生物合成的通路(包括PGC-1α),从而增加线粒体密度并提高氧化磷酸化效率[35]。这些效应对阿尔茨海默病和帕金森病等神经退行性疾病尤为有益,因为线粒体功能障碍通过损害神经元能量代谢和增加氧化损伤推动疾病进展[36-37]。
相比之下,植物性饮食(Plant-Based Diet, PBD)(如排除大部分或全部动物产品的素食)富含多酚类物质、抗氧化剂和膳食纤维,主要通过多重相互关联机制减少氧化应激和炎症,进而支持线粒体功能。白藜芦醇、槲皮素和表儿茶素等多酚类物质可通过激活AMP活化蛋白激酶(AMP-Activated Protein Kinase, AMPK)和上调去乙酰化酶(Sirtuins, SIRTs)来调节能量感应通路,这些酶类可调节代谢、应激抵抗和线粒体功能,从而激活PGC-1α,促进线粒体生物合成和氧化代谢,改善胰岛素敏感性并减少炎症细胞因子(图2)[38]。由图2可见,PBD和CR激活SIRT1,进而激活AMPK和PGC1α,导致线粒体生物合成增加、胰岛素信号增强。CR增强了氧化肌纤维中的线粒体融合。白藜芦醇、小檗碱和类黄酮等营养素通过激活SIRT1和AMPK增强线粒体生物合成。CR和这些营养物质通过降低ROS和炎症细胞因子水平共同有助于线粒体质量维持。另外,这些活性成分还能通过刺激线粒体自噬(通过自噬选择性清除受损或功能失调的线粒体)来增强线粒体质量控制,从而帮助维持健康且功能正常的线粒体网络。更重要的是,PBD的高抗氧化剂含量有助于中和ROS,保护线粒体DNA免受氧化损伤,并维持电子传递链的正常功能。亚麻籽、核桃等植物源的omega-3脂肪酸通过融入磷脂双分子层,维持线粒体膜完整性,优化流动性,提高电子传递效率[39]。此外,PBD中的高纤维含量促进了肠道微生物群的多样性,进而通过减少全身炎症和促进有益细菌的生长间接支持线粒体健康[40]。
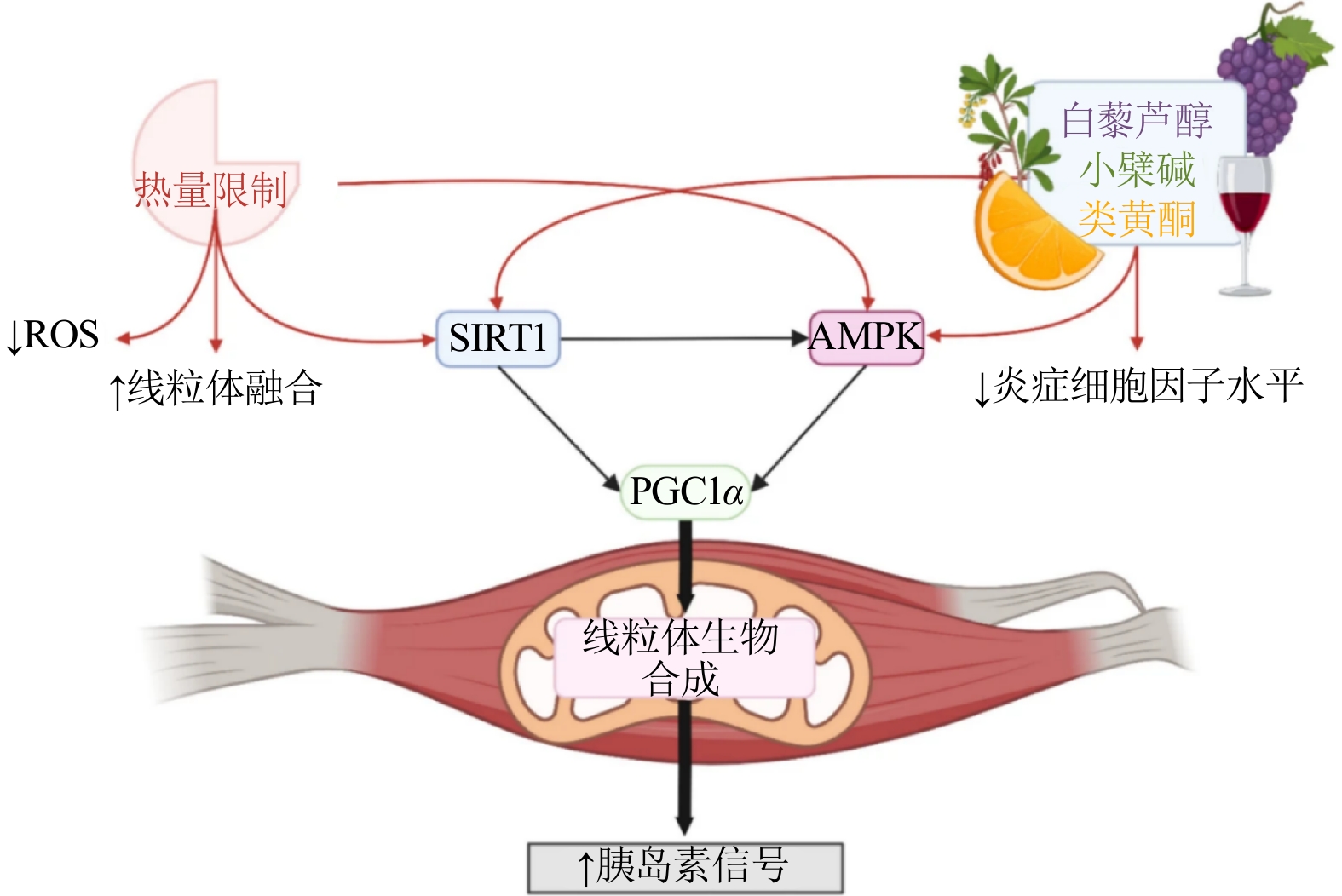 图 2 植物性饮食促进线粒体功能机制Fig. 2 Mechanisms of plant-based diets in enhancing mitochondrial function下载:
全尺寸图片
图 2 植物性饮食促进线粒体功能机制Fig. 2 Mechanisms of plant-based diets in enhancing mitochondrial function下载:
全尺寸图片
间歇性禁食(Intermittent Fasting, IF)和CR通过诱导增强线粒体适应性的细胞应激反应,在线粒体优化中发挥着关键作用。IF采用进食与禁食交替的模式,可能会使总体热量摄入适度减少,而CR是在不引起营养不良的情况下,保证每日持续减少热量摄入。这两种方式均能降低ROS水平,同时激活细胞和线粒体自噬,这对清除受损线粒体、促进线粒体更新至关重要(图2)[41-42]。由此带来的结果是线粒体质量和效率的整体提升,减少了功能失调细胞器的积累,而这些细胞器会促进衰老和代谢性疾病。此外,IF通过增加烟酰胺腺嘌呤二核苷酸(Nicotinamide Adenine Dinucleotide, NAD+)水平增强线粒体功能,进一步激活SIRTs。CR已被证明可上调PGC-1α,促进线粒体复制并转向更高效的氧化磷酸化。这些饮食策略还提高了代谢灵活性,使细胞能够根据能量供应情况更有效地在葡萄糖和脂肪酸代谢之间切换。因此,IF和CR与寿命延长、胰岛素敏感性提高和对T2D与肥胖等代谢性疾病的抵抗力增强有关[43]。此外,通过减少全身炎症和氧化应激,这两种饮食策略共同实现了对神经的保护,通过改善线粒体功能和增强突触可塑性来降低神经退行性疾病的风险。
除KD、PBD和禁食策略外,其他膳食模式也对优化线粒体功能起着关键作用。富含健康脂肪、抗氧化剂和多酚的地中海饮食,可通过激活AMPK和SIRT1信号通路,增强线粒体效率、防止氧化损伤并促进线粒体生物合成[44],其抗炎特性进一步支持神经保护和心血管健康。同样,限时进食(Time Restricted Eating, TRE)作为IF的一种形式,通过将进食窗口限制在特定时段(如8 h进食+16 h禁食),使线粒体能量代谢与昼夜节律同步,从而提升线粒体效率、细胞和线粒体自噬,同时提高胰岛素敏感性和代谢灵活性[45]。高蛋白饮食,尤其是富含亮氨酸等必需氨基酸的膳食,可通过哺乳动物雷帕霉素靶蛋白(mammalian Target of Rapamycin, mTOR)信号通路促进蛋白质合成,支持线粒体的维护与修复。均衡的高蛋白饮食对老年人和运动员来说尤其有益,它可以通过维持线粒体功能和肌肉质量,延缓肌肉减少症和减轻疲劳[46-47]。
4. 促进线粒体健康的运动方式
体育活动是调节线粒体健康的重要方式。有氧运动和无氧运动均已被证明可刺激线粒体生物合成、改善功能并支持质量控制机制。越来越多的证据表明,多种类型和强度的体育活动可以增强和维持线粒体功能,最终促进以能量代谢为核心的整体健康水平。
耐力训练,如中等强度的持续运动,是提升线粒体密度、氧化酶活性和脂肪酸氧化的最有效策略之一。这些适应变化主要由PGC-1α的上调驱动,PGC-1α通过启动一系列细胞事件促使线粒体生物合成增加[48]。规律性的耐力训练还会激活AMPK,有助于改善线粒体功能,并提高代谢效率。此外,它促进线粒体融合,支持线粒体的韧性和功能能力[49]。这些变化增强了胰岛素敏感性,提高了代谢灵活性,降低了代谢性疾病的发生风险。这些结果对T2D和肥胖等疾病患者尤其有益,因为线粒体功能障碍会损害葡萄糖摄取和能量调节[50]。耐力训练还减少了全身炎症和氧化应激,这两者都是线粒体退化的关键驱动因素。通过改善血管功能和毛细血管密度,进一步优化线粒体的氧气供应,增强细胞能量代谢。图3提供了关于运动如何增强线粒体健康的更详细和直观的解释,体现了线粒体合成、融合、裂变、线粒体自噬和蛋白质质量控制。运动可激活AMPK和SIRT1等信号通路,促进PGC-1α介导的核编码线粒体蛋白(NuGEMPs)的转录。运动诱导的ROS和钙信号进一步刺激线粒体合成。同时,线粒体融合(通过MFN1/2和OPA1)和裂变(通过DRP1和MFF)等动态过程可维持线粒体形态和功能。受损的线粒体通过线粒体自噬(由PINK1、Parkin、BNIP3和LC3介导)被选择性清除,而线粒体未折叠蛋白反应(Mitochondrial Unfolded Protein Response, UPRmt)通过泛素-蛋白酶体系统(Ubiquitin–Proteasome System, UPS)和自噬-溶酶体途径(Autophagy–Lysosomal Pathway, ALP)可维持蛋白质稳态。
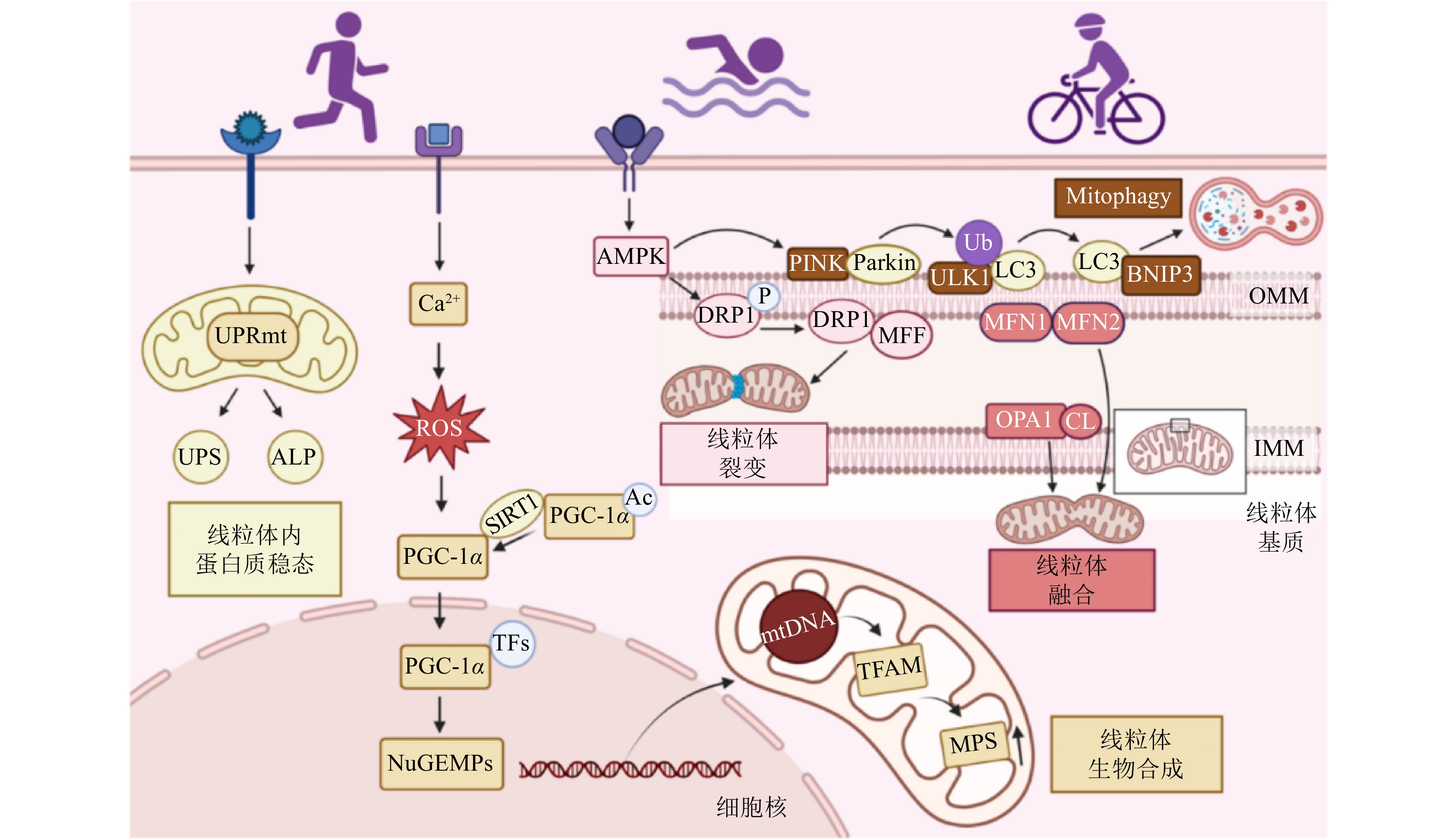 图 3 运动通过相互关联的过程增强线粒体健康Fig. 3 Exercise enhances mitochondrial health through interconnected processes下载:
全尺寸图片
图 3 运动通过相互关联的过程增强线粒体健康Fig. 3 Exercise enhances mitochondrial health through interconnected processes下载:
全尺寸图片
HIIT,尤其工作间隔时间较长(≥1 min)的训练方案,也成为诱导线粒体适应的强效刺激因素。HIIT通过交替进行短时间高强度训练和恢复,虽总时长较短,却能引发与耐力训练相当甚至更强的线粒体生物合成与呼吸功能增强[51-52]。与耐力训练相似,HIIT通过激活AMPK和PGC-1α信号通路,促使线粒体含量增加并提高代谢效率。这些适应变化有助于减少氧化应激和炎症,而这正是代谢和心血管疾病中线粒体功能障碍的核心特征。此外,HIIT对血管健康有多重益处:改善内皮功能、增加一氧化氮利用率、减少心肌细胞中的线粒体损伤[53]。因此,HIIT是改善代谢健康的一种高效且省时的训练方法,但由于其高强度特性,建议在专业指导下进行以确保安全。
抗阻训练传统上与肌肉肥大和力量发展相关,其在维持线粒体健康中也起着至关重要的作用。与主要通过代谢应激刺激线粒体适应的耐力训练和HIIT不同,抗阻训练对骨骼肌施加机械应力,激活与mTOR相关的独特信号通路。这些信号通路不仅支持肌肉蛋白质合成,还促进线粒体重塑[54]。已有证据表明抗阻训练可以增强肌肉质量和线粒体的数量及效率,这些适应性变化对于面临肌少症和线粒体衰退的老年人群尤其有益。此外,抗阻训练能降低线粒体碎片化和功能障碍的标志物,同时促进线粒体自噬,以选择性清除受损线粒体,从而保持线粒体的完整性[55-56]。这些改善有助于促进葡萄糖代谢、增强胰岛素敏感性,并降低代谢性疾病的风险[57]。此外,由抗阻训练引起的肌肉质量增加支持更高的静息代谢率和更强的脂肪酸氧化,可进一步改善能量平衡和代谢健康。
值得考虑的其他运动形式包括混合训练(Concurrent Training, CT)、低强度稳态训练(Low-Intensity Steady-State, LISS),以及瑜伽或太极。CT结合有氧运动和抗阻训练,能最佳地同时提高线粒体密度和肌肉力量,为老年人群和慢性病患者提供双重益处[58]。LISS(如长时间步行或骑行)能以最小的生理应激支持线粒体密度和脂肪氧化,特别适合体能较差者和慢性病患者[59]。瑜伽和太极等身心运动则可通过减少氧化应激、自主神经失衡和慢性炎症来改善线粒体效率,为慢性疲劳和神经退行性疾病等提供治疗潜能[60]。总之,这些多样化的运动方式共同提供了可扩展且易于实施的策略,以支持从运动员到代谢或神经系统疾病患者等广泛人群的线粒体健康。
5. 运动和饮食干预对线粒体健康的联合效应
众所周知,运动和饮食策略均可独立促进线粒体健康。新出现的证据表明,当两者结合时,可以通过重叠和独特的机制产生协同作用,效果优于单独使用任一方法[61]。具体而言,运动通过增加机体对ATP和代谢底物的需求来刺激线粒体更新,而饮食干预有助于改善底物利用率、优化氧化还原状态,进而增强线粒体功能能力。在高脂饮食诱导的肥胖动物模型中,运动结合饮食调整不仅能恢复线粒体含量,还可提高线粒体偶联效率,减少ROS的产生[62-63]。这种潜在的协同效应可能源于AMPK、SIRT1和PGC-1α等关键信号通路的放大激活,以及线粒体动力学(如裂变、融合和线粒体自噬)的改善[64]。
人体研究进一步证实了这些发现。一项对老年人的综合生活方式干预研究结果显示,与单独控制饮食或运动相比,二者结合的干预措施能更显著地改善骨骼肌线粒体呼吸功能和胰岛素敏感性[65]。有证据表明,将KD与耐力训练或抗阻训练结合可以增强线粒体脂肪酸氧化,同时降低氧化应激水平。将KD加入运动训练可以放大代谢适应,提高对脂肪酸氧化和酮体利用的依赖,从而进一步提高耐力表现和线粒体效率[66]。KD与抗阻训练结合,还可以通过保持线粒体完整性和功能以及肌肉质量来缓解衰老过程中的线粒体功能障碍[67]。相似的,在T2D患者中,综合饮食和运动计划相较于单一疗法,在线粒体酶活性和氧化磷酸化能力方面产生了更显著的提升[68]。
同样,富含多酚和omega-3脂肪酸的PBD与运动结合可以更有效地提高线粒体效率和代谢灵活性[69-70]。在运动的同时补充多酚可能通过激活AMPK和SIRT1信号通路进一步促进线粒体生物合成并减少氧化应激[69]。此外,TRE作为一种IF形式,能将营养供应与昼夜节律同步,从而在与运动结合时促进代谢适应。研究[71]表明,将TRE与耐力训练或抗阻训练相结合可以增强线粒体生物合成、脂肪氧化和细胞质量控制机制(如细胞和线粒体自噬)。
6. 增强线粒体健康的实用策略
综上所述,饮食与运动干预为通过协同互补机制,缓解线粒体功能障碍及降低慢性疾病风险提供了强有力的非药物策略。将上述基于循证的方案融入临床实践与日常生活,在改善代谢健康、增强神经保护、支持心血管功能及促进癌症预防方面展现出了巨大潜力。为将科学证据转化为实际行动,以下提出具体可实施的综合生活方式建议。
6.1 营养策略
(1)采用营养密集的全食物饮食:以富含抗氧化剂的蔬菜、水果、全谷物和健康脂肪为核心,为线粒体功能提供必需营养底物。
(2)增加多酚类食物的摄入:适当摄入含多酚的食物,如浆果、绿茶、黑巧克力和适量红酒,以刺激线粒体生物合成并减少氧化应激。
(3)保证Omega-3脂肪酸供给:通过亚麻籽、核桃和富含脂肪的鱼类等来源摄入omega-3脂肪酸,维护线粒体膜完整,优化能量生成效率。
(4)探索IF方案:通过IF或适当能量控制,激活自噬和线粒体自噬,促进功能失调线粒的体清除与更新。
(5)特定人群的KD:对于代谢或神经退行性疾病患者,在专业监督下采用KD可能增强线粒体效率并减少氧化应激。
(6)强化微量元素的补充:确保摄入足够的支持线粒体的微量营养素,如B族维生素(用于能量代谢)、镁(用于ATP生产)和辅酶Q10(用于电子传递链功能)。
(7)限制促炎食物的摄入:减少加工、添加糖和反式脂肪食物的摄入,这些与线粒体功能障碍和炎症相关。
6.2 运动策略
(1)进行耐力训练(如骑行、跑步或游泳),以增加线粒体密度并增强氧化能力。
(2)加入HIIT,以促进快速的线粒体适应并提高代谢灵活性,这对代谢综合征或肥胖患者尤其有益。
(3)每周进行至少2~3次抗阻训练,以支持线粒体质量控制,并减少与年龄相关的线粒体衰退。
(4)对于心血管疾病患者,结合有氧和抗阻训练,以改善线粒体功能和整体心脏健康。
(5)通过全天保持身体活动,减少久坐时间,因为长时间不活动会损害线粒体功能。
(6)平衡运动强度与充分恢复,以避免过度的氧化应激,这可能对线粒体健康产生负面影响。
6.3 其他生活方式策略
(1)优先考虑优质睡眠并与昼夜节律保持一致,因为睡眠中断会损害线粒体功能和能量代谢。
(2)通过正念练习、冥想或瑜伽管理压力,这些方法已被证明可以缓解与慢性压力相关的线粒体功能障碍。
(3)保持充足的水分摄入,因为适当的细胞水合作用支持线粒体效率和ATP生产。
(4)监测关键的代谢健康指标(如血糖、血脂、血压、体适能和肌肉力量),以评估营养和运动策略对线粒体功能的有效性。
7. 结论
线粒体功能障碍是代谢性疾病、神经退行性疾病、心血管疾病和癌症等慢性疾病发展的关键因素。线粒体生物合成受损、氧化磷酸化缺陷和过度的ROS产生会破坏细胞功能,从而推进促进疾病的发生发展。如,代谢性疾病中的线粒体效率低下会导致胰岛素抵抗和脂质积累,而线粒体碎片化和氧化损伤与神经退行性疾病相关。在心血管疾病中,线粒体功能障碍会加剧炎症反应并减少ATP生成,影响心脏功能,而癌细胞通常会重新编程线粒体以支持糖酵解,从而促进快速生长。在以能量为核心的健康理念框架内解决这些问题,即优先考虑细胞水平的能量代谢优化,可以为管理这些疾病提供更全面的方法。
有针对性的营养和运动策略对恢复线粒体健康和推进以能量为核心的健康理念至关重要。富含抗氧化剂、多酚、omega-3脂肪酸和关键微量营养素的营养密集型饮食,可减少氧化应激并增强线粒体功能。CR和KD可提高线粒体效率和灵活性,定期运动尤其是耐力训练、HIIT和抗阻训练可刺激线粒体生物合成并改善代谢健康。此外,充足的睡眠、压力管理和水分补充等生活方式因素,进一步支持线粒体的适应能力,为保持线粒体完整性提供了整体方法。
早期干预,特别是在儿童时期改善导致线粒体功能障碍的生活方式,对预防至关重要。随着研究的不断深入,开发评估线粒体功能的可靠方法,包括用于测量肌肉质量的生物阻抗分析,将有助于早期识别线粒体功能障碍,从而制定更有效、基于证据的策略以改善代谢健康并降低慢性疾病风险。最后,采用将线粒体健康与更广泛的生活方式改变相结合的以能量为核心的健康方法,可以使个人采取积极的措施改善代谢健康,并减少对药物或医疗干预的依赖。
-
图 1 多种细胞紊乱导致线粒体功能障碍在代谢性疾病的发展中起核心作用
Fig. 1 Various cellular disturbances contribute to mitochondrial dysfunction which plays a central role in the development of metabolic diseases
下载:
全尺寸图片
图 2 植物性饮食促进线粒体功能机制
Fig. 2 Mechanisms of plant-based diets in enhancing mitochondrial function
下载:
全尺寸图片
图 3 运动通过相互关联的过程增强线粒体健康
Fig. 3 Exercise enhances mitochondrial health through interconnected processes
下载:
全尺寸图片
表 1 与线粒体功能障碍相关的慢性疾病
Table 1 Chronic diseases associated with mitochondrial dysfunction
疾病类别 疾病名称 主要机制 代谢疾病 T2D 胰岛素敏感组织(如肌肉、肝脏)中的线粒体功能障碍会导致胰岛素抵抗和葡萄糖代谢紊乱 肥胖 脂肪组织和骨骼肌中的线粒体氧化能力下降,会导致脂肪堆积和代谢失衡 非酒精性脂肪肝 线粒体功能受损会破坏脂肪酸氧化过程,导致脂质在肝脏中积聚 神经退行性疾病 阿尔茨海默病 线粒体功能障碍会加剧氧化应激并损害能量代谢,进而导致神经元损伤和认知功能下降 帕金森病 线粒体复合体I的缺陷和ROS生成的增加与多巴胺能神经元的丢失有关 心血管
疾病心力衰竭 线粒体ATP生成的减少和氧化应激的加剧会损害心肌功能 动脉粥样硬化 血管细胞中的线粒体功能障碍会促进炎症和斑块形成 高血压 线粒体动力学受损和ROS的过量产生会导致血管功能障碍 其他 癌症 线粒体功能障碍可通过改变细胞代谢(如“瓦氏效应”)、增加ROS生成以及削弱细胞凋亡来促进肿瘤发生。与之相关的特定癌症包括乳腺癌、前列腺癌和结直肠癌,但线粒体的作用因癌症类型而异 慢性疲劳综合征(chronic fatigue syndrome) 有证据表明,有缺陷的线粒体能量生产可能是慢性疲劳综合征患者持续疲劳和身体机能下降的根本原因 衰老与年龄相关疾病 线粒体DNA突变和线粒体功能衰退是衰老过程的核心,会导致肌少症、骨质疏松和衰弱等年龄相关疾病 -
[1] BRAND M D, NICHOLLS D G. Assessing mitochondrial dysfunction in cellsp[J]. Biochemical Journal, 2011, 435(2):297-312. doi: 10.1042/BJ20110162 [2] WALLACE D C. Mitochondrial genetic medicine[J]. Nature Genetics, 2018, 50(12):1642-1649. doi: 10.1038/s41588-018-0264-z [3] PICARD M, MCEWEN B S, EPEL E S, et al. An energetic view of stress: Focus on mitochondria[J]. Frontiers in Neuroendocrinology, 2018, 49:72-85. doi: 10.1016/j.yfrne.2018.01.001 [4] MURPHY M P, HARTLEY R C. Mitochondria as a therapeutic target for common pathologies[J]. Nature Reviews Drug Discovery, 2018, 17(12):865-886. doi: 10.1038/nrd.2018.174 [5] JANG J Y, BLUM A, LIU J, et al. The role of mitochondria in aging[J]. Journal of Clinical Investigations, 2018, 128(9):3662-3670. doi: 10.1172/JCI120842 [6] ANDERSON E J, KYPSON A P, RODRIGUEZ E, et al. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart[J]. Journal of the American College of Cardiology, 2009, 54(20):1891-1898. doi: 10.1016/j.jacc.2009.07.031 [7] GANO L B, PATEL M, RHO J M. Ketogenic diets, mitochondria, and neurological diseases[J]. Journal of Lipid Research, 2014, 55(11):2211-2228. doi: 10.1194/jlr.R048975 [8] HOLLOSZY J O. Regulation of mitochondrial biogenesis and GLUT4 expression by exercise[J]. Comprehensive Physiology, 2011, 1(2):921-940. doi: 10.1002/j.2040-4603.2011.tb00346.x [9] SCARPULLA R C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network[J]. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2011, 1813(7):1269-1278. doi: 10.1016/j.bbamcr.2010.09.019 [10] MOOTHA V K, LINDGREN C M, ERIKSSON K F, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes[J]. Nature Genetics, 2003, 34(3):267-273. doi: 10.1038/ng1180 [11] LOWELL B B, SHULMAN G I. Mitochondrial dysfunction and type 2 diabetes[J]. Science, 2005, 307(5708):384-387. doi: 10.1126/science.1104343 [12] PETERSEN K F, DUFOUR S, BEFROY D, et al. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes[J]. The New England Journal of Medicine, 2004, 350(7):664-671. doi: 10.1056/NEJMoa031314 [13] HOEHN K L, SALMON A B, HOHNEN-BEHRENS C, et al. Insulin resistance is a cellular antioxidant defense mechanism[J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(42):17787-17792. [14] KELLEY D E, HE J, MENSHIKOVA E V, et al. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes[J]. Diabetes, 2002, 51(10):2944-2950. doi: 10.2337/diabetes.51.10.2944 [15] BHATTI J S, BHATTI G K, REDDY P H. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies[J]. Biochimica et Biophysica Acta Molecular Basis of Disease, 2017, 1863(5):1066-1077. doi: 10.1016/j.bbadis.2016.11.010 [16] SANYAL A J, CAMPBELL–SARGENT C, MIRSHAHI F, et al. Nonalcoholic steatohepatitis:Association of insulin resistance and mitochondrial abnormalities[J]. Gastroenterology, 2001, 120(5):1183-1192. doi: 10.1053/gast.2001.23256 [17] PESSAYRE D, FROMENTY B. NASH: A mitochondrial disease[J]. Journal of Hepatology, 2005, 42(6):928-940. doi: 10.1016/j.jhep.2005.03.004 [18] SWERDLOW R H, KHAN S M. A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease[J]. Medical Hypotheses, 2004, 63(1):8-20. doi: 10.1016/j.mehy.2003.12.045 [19] REDDY P H, MAO P Z, MANCZAK M. Mitochondrial structural and functional dynamics in Huntington's disease[J]. Brain Research Reviews, 2009, 61(1):33-48. doi: 10.1016/j.brainresrev.2009.04.001 [20] TIMPKA J, CENCI M.A, ODIN P.Etiology and pathogenesis of Parkinson's disease[M]//Movement Disorders Curricula.Vienna:Springer Vienna, 2017:95-101. [21] EXNER N, LUTZ A K, HAASS C, et al. Mitochondrial dysfunction in Parkinson's disease: Molecular mechanisms and pathophysiological consequences[J]. The EMBO Journal, 2012, 31(14):3038-3062. doi: 10.1038/emboj.2012.170 [22] PICKRELL A M, YOULE R J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease[J]. Neuron, 2015, 85(2):257-273. doi: 10.1016/j.neuron.2014.12.007 [23] COZZOLINO M, CARRÌ M T. Mitochondrial dysfunction in ALS[J]. Progress in Neurobiology, 2012, 97(2):54-66. doi: 10.1016/j.pneurobio.2011.06.003 [24] MANFREDI G, XU Z S. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS[J]. Mitochondrion, 2005, 5(2):77-87. doi: 10.1016/j.mito.2005.01.002 [25] SMITH R A, MURPHY M P. Mitochondria-targeted antioxidants as therapies[J]. Discovery Medicine, 2011, 11(57):106-114. [26] GUSTAFSSON A B, GOTTLIEB R A. Heart mitochondria: Gates of life and death[J]. Cardiovascular Research, 2008, 77(2):334-343. [27] DIKALOV S I, UNGVARI Z. Role of mitochondrial oxidative stress in hypertension[J]. American Journal of Physiology Heart and Circulatory Physiology, 2013, 305(10):1417-1427. doi: 10.1152/ajpheart.00089.2013 [28] BALLINGER S W, PATTERSON C, KNIGHT-LOZANO C A, et al. Mitochondrial integrity and function in atherogenesis[J]. Circulation, 2002, 106(5):544-549. doi: 10.1161/01.CIR.0000023921.93743.89 [29] WALLACE D C. Mitochondria and cancer[J]. Nature Reviews Cancer, 2012, 12(10):685-698. doi: 10.1038/nrc3365 [30] CHATTERJEE A, MAMBO E, SIDRANSKY D. Mitochondrial DNA mutations in human cancer[J]. Oncogene, 2006, 25(34):4663-4674. doi: 10.1038/sj.onc.1209604 [31] XIE L L, SHI F, TAN Z Q, et al. Mitochondrial network structure homeostasis and cell death[J]. Cancer Science, 2018, 109(12):3686-3694. doi: 10.1111/cas.13830 [32] SENA L A, CHANDEL N S. Physiological roles of mitochondrial reactive oxygen species[J]. Molecular Cell, 2012, 48(2):158-167. doi: 10.1016/j.molcel.2012.09.025 [33] VYAS S, ZAGANJOR E, HAIGIS M C. Mitochondria and cancer[J]. Cell, 2016, 166(3):555-566. doi: 10.1016/j.cell.2016.07.002 [34] MEANS C M D, MEANS C.Good energy:The surprising connection between metabolism and limitless health[M].New York:Avery, 2004. [35] KIM S, PARK D H, MOON S, et al. Ketogenic diet with aerobic exercise can induce fat browning:Potential roles of β-hydroxybutyrate[J]. Frontiers in Nutrition, 2024, 11:1443483. doi: 10.3389/fnut.2024.1443483 [36] NEWMAN J C, VERDIN E. Ketone bodies as signaling metabolites[J]. Trends in Endocrinology & Metabolism, 2014, 25(1):42-52. [37] CHENG C W, ADAMS G B, PERIN L, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression[J]. Cell Stem Cell, 2014, 14(6):810-823. doi: 10.1016/j.stem.2014.04.014 [38] LAGOUGE M, ARGMANN C, GERHART-HINES Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α[J]. Cell, 2006, 127(6):1109-1122. doi: 10.1016/j.cell.2006.11.013 [39] LANZA I R, BLACHNIO-ZABIELSKA A, JOHNSON M L, et al. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet[J]. American Journal of Physiology Endocrinology and Metabolism, 2013, 304(12):E1391-E1403. doi: 10.1152/ajpendo.00584.2012 [40] MANN E R, LAM Y K, UHLIG H H. Short-chain fatty acids:Linking diet, the microbiome and immunity[J]. Nature Reviews Immunology, 2024, 24(8):577-595. doi: 10.1038/s41577-024-01014-8 [41] LÓPEZ-LLUCH G, HUNT N, JONES B, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency[J]. Proceedings of the National Academy of Sciences of the United States of America, 2006, 103(6):1768-1773. [42] ZHANG A R, WANG J Y, ZHAO Y N, et al. Intermittent fasting, fatty acid metabolism reprogramming, and neuroimmuno microenvironment: Mechanisms and application prospects[J]. Frontiers in Nutrition, 2024, 11:1485632. doi: 10.3389/fnut.2024.1485632 [43] CANTÓ C, AUWERX J. Caloric restriction, SIRT1 and longevity[J]. Trends in Endocrinology & Metabolism, 2009, 20(7):325-331. [44] ESTRUCH R, ROS E, SALAS-SALVADÓ J, et al. Primary prevention of cardiovascular disease with a Mediterranean diet[J]. The New England Journal of Medicine, 2013, 368(14):1279-1290. doi: 10.1056/NEJMoa1200303 [45] CHAIX A, ZARRINPAR A, MIU P, et al. Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges[J]. Cell Metabolism, 2014, 20(6):991-1005. doi: 10.1016/j.cmet.2014.11.001 [46] WALRAND S, SHORT K R, BIGELOW M L, et al. Functional impact of high protein intake on healthy elderly people[J]. American Journal of Physiology Endocrinology and Metabolism, 2008, 295(4):E921-E928. doi: 10.1152/ajpendo.90536.2008 [47] BURTSCHER J, STRASSER B, BURTSCHER M. A mito-centric view on muscle aging and function[J]. Frontiers in Public Health, 2023, 11:1330131. [48] WU Z, PUIGSERVER P, ANDERSSON U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1[J]. Cell, 1999, 98(1):115-124. doi: 10.1016/S0092-8674(00)80611-X [49] LITTLE J P, SAFDAR A, CERMAK N, et al. Acute endurance exercise increases the nuclear abundance of PGC-1alpha in trained human skeletal muscle[J]. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 2010, 298(4):R912-R917. doi: 10.1152/ajpregu.00409.2009 [50] HOLLOSZY J O. Skeletal muscle "mitochondrial deficiency" does not mediate insulin resistance[J]. The American Journal of Clinical Nutrition, 2009, 89(1):463S-466S. doi: 10.3945/ajcn.2008.26717C [51] GIBALA M J, LITTLE J P, VAN ESSEN M, et al. Short-term sprint interval versus traditional endurance training:Similar initial adaptations in human skeletal muscle and exercise performance[J]. The Journal of Physiology, 2006, 575(3):901-911. doi: 10.1113/jphysiol.2006.112094 [52] MACINNIS M J, ZACHAREWICZ E, MARTIN B J, et al. Superior mitochondrial adaptations in human skeletal muscle after interval compared to continuous single-leg cycling matched for total work[J]. The Journal of Physiology, 2017, 595(9):2955-2968. doi: 10.1113/JP272570 [53] IQBAL S, HOOD D A. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts[J]. American Journal of Physiology Cell Physiology, 2014, 306(12):C1176-C1183. doi: 10.1152/ajpcell.00017.2014 [54] PORTER C, REIDY P T, BHATTARAI N, et al. Resistance exercise training alters mitochondrial function in human skeletal muscle[J]. Medicine and Science in Sports and Exercise, 2015, 47(9):1922-1931. doi: 10.1249/MSS.0000000000000605 [55] GUO C, WU R Y, DOU J H, et al. Mitophagy-dependent cardioprotection of resistance training on heart failure[J]. Journal of Applied Physiology, 2023, 135(6):1390-1401. doi: 10.1152/japplphysiol.00674.2023 [56] PARRY H A, ROBERTS M D, KAVAZIS A N. Human skeletal muscle mitochondrial adaptations following resistance exercise training[J]. International Journal of Sports Medicine, 2020, 41(6):349-359. doi: 10.1055/a-1121-7851 [57] HOOD D A, MEMME J M, OLIVEIRA A N, et al. Maintenance of skeletal muscle mitochondria in health, exercise, and aging[J]. Annual Review of Physiology, 2019, 81:19-41. doi: 10.1146/annurev-physiol-020518-114310 [58] KONOPKA A R, HARBER M P. Skeletal muscle hypertrophy after aerobic exercise training[J]. Exercise and Sport Sciences Reviews, 2014, 42(2):53-61. doi: 10.1249/JES.0000000000000007 [59] HELGERUD J, HØYDAL K, WANG E, et al. Aerobic high-intensity intervals improve VO2max more than moderate training[J]. Medicine and Science in Sports and Exercise, 2007, 39(4):665-671. doi: 10.1249/mss.0b013e3180304570 [60] BURIC I, FARIAS M, JONG J, et al. What is the molecular signature of mind-body interventions? A systematic review of gene expression changes induced by meditation and related practices[J]. Frontiers in Immunology, 2017, 8:670. doi: 10.3389/fimmu.2017.00670 [61] KONOPKA A R, NAIR K S. Mitochondrial and skeletal muscle health with advancing age[J]. Molecular and Cellular Endocrinology, 2013, 379(1-2):19-29. doi: 10.1016/j.mce.2013.05.008 [62] PALMER N O, BAKOS H W, OWENS J A, et al. Diet and exercise in an obese mouse fed a high-fat diet improve metabolic health and reverse perturbed sperm function[J]. American Journal of Physiology Endocrinology and Metabolism, 2012, 302(7):E768-E780. doi: 10.1152/ajpendo.00401.2011 [63] SILVESTRI E, GIACCO A. Diet, exercise, and the metabolic syndrome:Enrollment of the mitochondrial machinery[J]. Nutrients, 2022, 14(21):4519. doi: 10.3390/nu14214519 [64] PALIKARAS K, LIONAKI E, TAVERNARAKIS N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology[J]. Nature Cell Biology, 2018, 20(9):1013-1022. doi: 10.1038/s41556-018-0176-2 [65] ROBINSON M M, DASARI S, KONOPKA A R, et al. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans[J]. Cell Metabolism, 2017, 25(3):581-592. doi: 10.1016/j.cmet.2017.02.009 [66] VOLEK J S, NOAKES T, PHINNEY S D. Rethinking fat as a fuel for endurance exercise[J]. European Journal of Sport Science, 2015, 15(1):13-20. doi: 10.1080/17461391.2014.959564 [67] MCSWINEY F T, WARDROP B, HYDE P N, et al. Keto-adaptation enhances exercise performance and body composition responses to training in endurance athletes[J]. Metabolism, 2018, 81:25-34. doi: 10.1016/j.metabol.2017.10.010 [68] MENSHIKOVA E V, RITOV V B, FAIRFULL L, et al. Effects of exercise on mitochondrial content and function in aging human skeletal muscle[J]. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences, 2006, 61(6):534-540. doi: 10.1093/gerona/61.6.534 [69] LIU Y, FANG M L, TU X H, et al. Dietary polyphenols as anti-aging agents:Targeting the hallmarks of aging[J]. Nutrients, 2024, 16(19):3305. doi: 10.3390/nu16193305 [70] MCGLORY C, CALDER P C, NUNES E A. The influence of omega-3 fatty acids on skeletal muscle protein turnover in health, disuse, and disease[J]. Frontiers in Nutrition, 2019, 6:144. doi: 10.3389/fnut.2019.00144 [71] ANTONI R, JOHNSTON K, COLLINS A, et al. The effects of intermittent energy restriction on indices of cardiometabolic health[J]. Research in Endocrinology, 2014:1-24.